

Introduction
Form 25 is the licence issued under the Drugs and Cosmetics Rules, 1945 for the manufacture for sale or distribution of drugs, excluding:
Form 25F is the licence issued under the Drugs and Cosmetics Rules, 1945 for the manufacture of drugs specified in Schedule X for the purpose of sale or distribution. Schedule X includes stringent-control narcotic and psychotropic drugs that carry a higher potential for abuse or dependence.
Conditions for the grant of a licence in Form 25 or Form 25F (Rule 71)
Before a licence in Form 25 or Form 25F is granted the following conditions shall be complied with by the applicant:
(1) the manufacture shall be conducted under the active direction and personal supervision of competent technical staff consisting at least of one person who is a whole time employee and who is—
(a) a graduate in Pharmacy or Pharmaceutical Chemistry of a University established in India by law or has an equivalent qualification recognized and notified by the Central Government for such purpose and has had at least eighteen months’ practical experience after the graduation in the manufacture of drugs. This period of experience may, however, be reduced by six months if the person has undergone training in manufacture of drugs for a period of six months during his University course; or
(b) a graduate in Science of a University established in India by law or has an equivalent qualification recognised and notified by the Central Government for such purpose who for the purpose of his degree has studied Chemistry as a principal subject and has had at least three years’ practical experience in the manufacture of drugs after his graduation; or
(c) a graduate in Chemical Engineering or Chemical Technology or Medicine of a University established in India by law or has an equivalent qualification recognised and notified by the Central Government for such purpose with general training and practical experience, extending over a period of not less than three years in the manufacture of drugs, after his graduation; or
(d) holding any foreign qualification the quality and content of training of which are comparable with those prescribed in clause(a), clause (b) or clause (c) and is permitted to work as competent technical staff under this rule by the Central Government:
Provided that any person who was immediately before the 29th June, 1957, actively directing and personally supervising the manufacture of drugs and whose name was accordingly entered in any licence granted in Form 25 or Form 25F as it existed before that date shall be deemed to be qualified for the purposes of this rule:
Provided further that for drugs other than those specified in Schedules C, C(l), and X and meant for veterinary use, the wholetime employee under whose supervision the manufacture is conducted shall be a graduate in Veterinary.
Science or Pharmacy or General Science or Medicine of a University recognized by the Central Government and who has had atleast three years’ practical experience in the manufacture of drugs excluding graduate in Pharmacy who shall have at least eighteen months’ practical experience in the manufacture of drugs:
Provided also that the licensing authority may, in the matter of manufacture of disinfectant fluid insecticides, liquid paraffin, medicinal gases, non-chemical contraceptives, plaster of paris and surgical dressings, for the manufacture of which the knowledge of Pharmaceutical Chemistry or Pharmacy is not essential, permit the manufacture of the substance under the active direction and personal supervision of the competent technical staff, who, although not having any of the qualifications included in clause (a), (b) or (c) of this rule, has, in the opinion of the licensing authority, adequate experience in the manufacture of such substance.

(2) The factory premises shall comply with the conditions prescribed in Schedule M.
(3) The applicant shall provide adequate space, plant and equipment for the manufacturing operations; the space, plant and equipment recommended for various operations are given in Schedule M.
(4) The applicant shall provide and maintain adequate staff, premises and laboratory equipment for carrying out tests of the strength, quality and purity of the substances at the testing unit which shall be separate from the manufacturing unit and head of the testing unit shall be independent of the head of the manufacturing unit:
Provided that the manufacturing units, which, before the commencement of the Drugs and Cosmetics (Amendment) Rules, 1977, were making arrangements with institutions approved by the licensing authority for such tests to be carried out on their behalf may continue such arrangements up to the 30th June, 1977:
Provided further that for tests requiring sophisticated instrumentation techniques or biological or microbiological methods other than sterility the licensing authority may permit such tests to be conducted by institutions approved by it under Part XV (A) of these rules for this purpose.
(4A) The head of the testing unit referred to in condition (4) shall possess a degree in Medicine or Science or Pharmacy or Pharmaceutical Chemistry of a University recognised for this purpose and shall have experience in the testing of drugs, which in the opinion of the licensing authority is considered adequate.
(5) The applicant shall make adequate arrangements for the storage of drugs manufactured by him.
(6) The applicant shall, while applying for a licence to manufacture drugs, furnish to the licensing authority evidence and date justifying that the drugs—
(i) contain the constituent ingredients in therapeutic/prophylactic quantities as determined in relation to the claims or conditions for which the medicines are recommended for use or claimed to be useful;
(ii) are safe for use in the context of the vehicles, excipients additives and pharmaceutical aids used in the formulation and under the conditions in which the formulations for administration and use are recommended;
(iii) are stable under the conditions of storage recommended; and
(iv) contain such ingredients and in such quantities for which there is therapeutic justification.
(v) have the approval, in writing, in favour of the applicant to manufacture drug formulations falling under the purview of new drug as defined in rule 122E, from the licensing authority as defined in clause (b) of rule 21.
(7) The licensee shall comply with the requirements of ‘Good Manufacturing Practices’ as laid down in Schedule M.
(8) The applicant shall make application for grant of licence for a drug formulation containing single active ingredient only in proper name.
(9) In case the applicant intends to market the drug under a brand name or trade name, the applicant shall furnish an undertaking in Form 51 to the licensing authority to the effect that to the best of his knowledge based on search in trade marks registry, central data base for brand name or trade name of drugs maintained by Central Drugs Standard Control Organisation, literature and reference books on details of drug formulations in India, and internet, such or similar brand name or trade name is not already in existence with respect to any drug in the country and the proposed brand name or trade name shall not lead to any confusion or deception in the market.

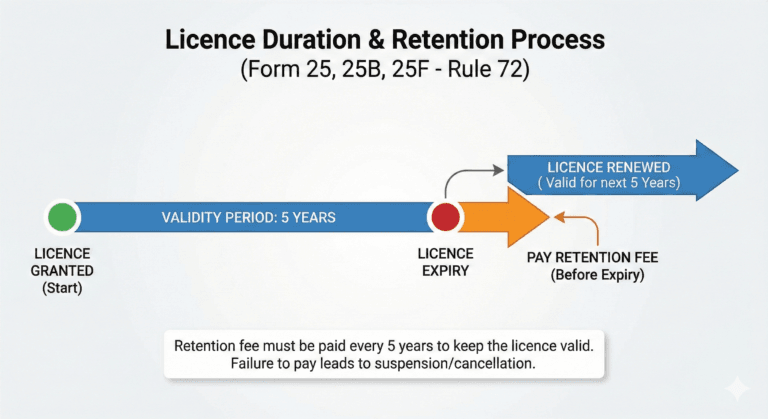
Duration of licence (Rule 72)
(1) A licence issued in Form 25, Form 25B and Form 25F shall remain valid if the licencee deposits a licence retention referred to in sub-rule (2) before the expiry of a period of every succeeding five years from the date of its issue, unless, it is suspended or cancelled by the licensing authority.
Conditions of licence in Form 25 and Form 25F (Rule 74)
A licence in Form25 and Form 25F shall be subject to the conditions stated therein and to the following further conditions, namely:—
A. Accounts of the drugs specified in Schedule X used for the manufacture—
B. Accounts of production—
C. Accounts of the manufactured drugs—
Disclaimer: The information contained in this Article is intended solely for personal non-commercial use of the user who accepts full responsibility of its use. The information in the article is general in nature and should not be considered to be legal, tax, accounting, consulting or any other professional advice. We make no representation or warranty of any kind, express or implied regarding the accuracy, adequacy, reliability or completeness of any information on our page/article.